|
Sistema nervioso > Antiepilépticos > Antiepilépticos > Otros antiepilépticos Mecanismo de acciónBrivaracetam Brivaracetam muestra una alta y selectiva afinidad por la proteína 2A de la vesícula sináptica (SV2A), una glicoproteína transmembrana encontrada a nivel presináptico en las neuronas y en células endocrinas. Aunque el papel exacto de esta proteína todavía tiene que ser dilucidado, se ha visto que modula la exocitosis de los neurotransmisores. Se cree que la unión a SV2A es el mecanismo principal de la actividad anticonvulsivante de brivaracetam. Indicaciones terapéuticasBrivaracetamComo terapia concomitante en el tto. de las crisis de inicio parcial con o sin generalización secundaria en ads. y adolescentes y niños a partir de 2 años con epilepsia. PosologíaBrivaracetam
Para acceder a la información de posología en Vademecum.es debes conectarte con tu email y clave o registrarte. Conéctate Regístrate Vía oral o IV: Vía oral. Comprimidos: administrar enteros, con algo de líquido, pueden tomarse con o sin alimentos. Solución oral puede diluirse en agua o zumo poco antes de su administración y se puede tomar con o sin alimentos. La solución oral puede utilizarse una sonda nasogástrica o una sonda de gastrostomía. Hipersensibilidad a brivaracetam, a otros derivados de la pirrolidona. Advertencias y precaucionesBrivaracetamI.H., niños< 4 años, no se ha establecido todavía la seguridad y eficacia. Riesgo de pensamientos y comportamientos suicidas, monitorizar para detectar signos de pensamientos y comportamientos suicidas y considerar el tto. adecuado. Enf.renal en fase final sometidos a diálisis no se recomienda debido a la falta de datos. Insuficiencia hepáticaBrivaracetamPrecaución. Los datos clínicos sobre el uso de brivaracetam en pacientes con I.H. preexistente son escasos. Se recomienda ajuste de dosis en I.H. Dosis de inicio recomendada para ads., adolescentes y niños > 4 años con más de 50 kg: 50 mg/día. I.H. dosis máx: 150 mg/día dividida en 2 dosis. Niños a partir de 4 años y adolescentes con menos de 50 kg: dosis de inicio: 1 mg/kg/día. La dosis máxima no debe superar los 3 mg/kg/día. No hay datos clínicos disponibles en los pacientes pediátricos con I.H. Insuficiencia renalBrivaracetamNo se require ajuste de dosis en pacientes con I.R. No está recomendado en enfermedad renal en fase final sometidos a diálisis debido a la falta de datos. InteraccionesBrivaracetamConcentraciones plasmáticas aumentadas por: inhibidores potentes del CYP2C19 (ej. fluconazol, fluvoxamina), pero el riesgo de una interacción mediada por el CYP2C19 clínicamente relevante se considera bajo. Hay escasos datos sobre el uso de brivaracetam en mujeres embarazadas. No hay datos de la tranferencia placentaria en humanos, pero en ratas, brivaracetam mostró que atravesiesa la placenta fácilmente. Se desconoce el posible riesgo en humanos. Los estudios en animales no detectaron ningún potencial efecto teratogénico de brivaracetam. En ensayos clínicos, se utilizó brivaracetam como tratamiento concomitante, y cuando se utilizó con carbamazepina, indujo un aumento relacionado con la dosis en la concentración del metabolito activo carbamazepina-epóxido. No hay datos suficientes para determinar la importancia clínica de este efecto en el embarazo. Como medida de precaución, no se debe utilizar brivaracetam durante el embarazo a no ser que sea clínicamente necesario (ej. si el beneficio para la madre supera el posible riesgo para el feto). LactanciaBrivaracetamSe desconoce si brivaracetam se excreta en la leche materna. Estudios en ratas mostraron excreción de brivaracetam en la leche. Se debe decidir si es necesario interrumpir la lactancia o interrumpir el tratamiento con brivaracetam tras consider el beneficio del medicamento para la madre. En caso de administración conjunta de brivaracetam y carbamazepina, la cantidad de carbamazepina-epóxido excretada en la leche materna puede aumentar. No hay datos suficientes para determinar la importancia clínica. Efectos sobre la capacidad de conducirBrivaracetamLa influencia de brivaracetam sobre la capacidad para conducir y utilizar máquinas es pequeña o moderada. Debido a las posibles diferencias de sensibilidad individual, algunos pueden experimentar somnolencia, mareo y otros síntomas relacionados con el SNC. Se debe advertir a los pacientes que no conduzcan ni manejen máquinas potencialmente peligrosas hasta que se hayan familiarizado con los efectos de brivaracetam sobre su capacidad para realizar dichas actividades. Reacciones adversasBrivaracetamGripe; disminución del apetito; depresión, ansiedad, insomnio, irritabilidad; mareo, somnolencia, convulsión, vértigo; infecciones del tracto respiratorio superior, tos; náuseas, vómitos, estreñimiento; fatiga © Vidal Vademecum Fuente: El contenido de esta monografía de principio activo según la clasificación ATC, ha sido redactado teniendo en cuenta la información clínica de todos los medicamentos autorizados y comercializados en España clasificados en dicho código ATC. Para conocer con detalle la información autorizada por la AEMPS para cada medicamento, deberá consultar la correspondiente Ficha Técnica autorizada por la AEMPS. Monografías Principio Activo: 21/07/2022
1. DENOMINACIÓN DISTINTIVA:
Briviact®
Brivaracetam 3. FORMA FARMACÉUTICA Y FORMULACIÓN
Tableta Fórmula.Cada tableta contiene: Brivaracetam 10 mg 25 mg 50 mg 75 mg 100 mg Excipiente cbp Tableta Tableta Tableta Tableta Tableta 4. INDICACIONES TERAPÉUTICAS
Briviact® está indicada como monoterapia y terapia de adición en el tratamiento de las crisis de inicio parcial con o sin generalización secundaria en pacientes con epilepsia de 4 años de edad o mayores. 5. FARMACOCINÉTICA Y FARMACODINAMIA
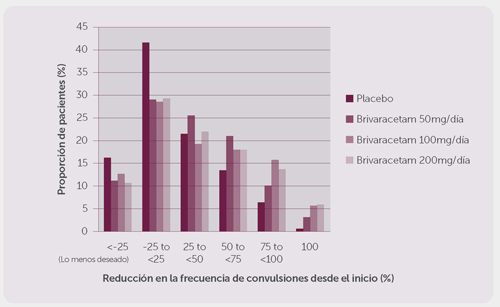
Farmacocinética: Brivaracetam exhibe una farmacocinética lineal e independiente de tiempo con baja variabilidad intra y entre sujetos, asimismo presenta una absorción completa, muy baja unión a proteínas, excreción renal después de una biotransformación extensiva, y metabolitos inactivos a nivel farmacológico. Brivaracetam tiene un bajo potencial de interacción entre fármacos. La farmacocinética de brivaracetam es similar cuando se usa como monoterapia o como terapia de adición para el tratamiento de las crisis de inicio parcial. Absorción La administración conjunta con alimentos altos en grasa retarda la tasa de absorción del medicamento mientras que el alcance de absorción permaneció sin cambios. Distribución Debido a su favorable lipofilicidad (Log P), que resulta en la alta permeabilidad de la membrana celular, el brivaracetam penetra rápidamente en el cerebro. Este medicamento se distribuye rápida y uniformemente en la mayoría de los tejidos. En roedores, la proporción de la concentración cerebro a plasma se equilibra con rapidez, indicando la rápida penetración en el cerebro y se acerca a 1, indicando la ausencia del transporte activo. Metabolismo Eliminación Poblaciones especiales Ancianos Insuficiencia renal Insuficiencia hepática Población pediátrica Género Raza Farmacodinamia: La sustancia activa es (2S)-2-[(4R)-2-oxo-4-propiltetrahidro-1H-pirrol-1-ilo] butanamida. Mecanismo de acción Los datos farmacodinámicos preclínicos de brivaracetam sugieren una alta potencia y eficacia en la supresión de crisis en diversos tipos de epilepsia en animales incluyendo crisis parciales y generalizadas, así como contra el estado epiléptico y mioclónico. Interacciones con el alcohol Abuso potencial Dependencia Efectos en el intervalo QT Frecuencia de las crisis epilépticas Estudios clínicos. La dosis diaria del brivaracetam varió de 5 a 200 mg/día a través de estos estudios. Todos los estudios tuvieron un período inicial de ocho semanas seguido por un período de tratamiento de 12 semanas sin titulación ascendente. Un número de 1,558 pacientes recibió el fármaco del estudio de los cuales, 1,099 recibieron brivaracetam. El criterio de reclutamiento de los estudios requirió que los pacientes tuvieran crisis de inicio parcial no controlado con uno o dos antiepilépticos concomitantes. Se requirió que los pacientes tuvieran, por lo menos ocho crisis de inicio parcial durante el período inicial. Los antiepilépticos tomados con mayor frecuencia en el período del registro del estudio fueron carbamazepina (40.6%), lamotrigina (25.2%), valproato (20.5%), oxcarbazepina (16.0%), topiramato (13.5%), fenitoína (10.2%) y levetiracetam (9.8%). En el estudio N01358, el 18.9% de los sujetos tenía un historial de 0-1 antiepiléptico previo, un 33.8% de 2 - 4 antiepilépticos previos, y el 47.2% ≥ 5 antiepilépticos previos. La frecuencia promedio inicial de crisis a través de los tres estudios fue de 9 por 28 días. Los pacientes habían tenido una duración promedio de epilepsia de aproximadamente 23 años. Las valoraciones de la eficacia se resumen en la Tabla 2. En general, el brivaracetam fue eficaz para el tratamiento adjunto de las crisis de inicio parcial con o sin generalización secundaria en pacientes de 16 años de edad y mayores, entre 50 y 200 mg/día. Los resultados del porcentaje de reducción sobre el placebo del N01252 y del N01253 se basan en la frecuencia de las crisis de inicio parcial por 28 días para permitir la comparación de los resultados presentados en el N01358, aunque los análisis sobre la eficacia primaria del N01252 y del N01253 se basaron en la frecuencia de crisis de inicio parcial por siete días. Las conclusiones respecto a la importancia estadística no se impactaron por el cambio de la duración en la que la frecuencia de la crisis de inicio parcial se estandarizó para los estudios N01252 y N01253. Tabla 2: Valoraciones Clave de la Eficacia respecto a la Frecuencia de crisis de inicio parcial por 28 días. Estudio Placebo Brivaracetam* Significativos a nivel estadístico (valor p) 50 mg/día 100 mg/día 200 mg/día Estudio N01253(1) n = 96 n = 101 Porcentaje de reducción sobre el placebo (%) NA 22.0* (p=0.004) ~ ~ Tasa de respuesta**(%) 16.7 32.7* (p=0.008) ~ ~ Porcentaje de reducción promedio desde el inicio (%) 17.8 30.5* (p=0.003) ~ ~ Estudio N01252(1) n = 100 n = 99 n = 100 Porcentaje de reducción sobre el placebo (%) NA 9.2 (p=0.274) 20.5(2) (p=0.010) ~ Tasa de respuesta** (%) 20.0 27.3 (p=0.372) 36.0(2) (p=0.023) ~ Porcentaje de reducción promedio desde el inicio (%) 17.0 26.8 (p=0.092) 32.5(2) (p=0.004) ~ Estudio N01358 n = 259 n = 252 n = 249 Porcentaje de reducción sobre el placebo (%) NA ~ 22.8* (p<0.001) 23.2* (p<0.001) Tasa de respuesta** (%) 21.6 ~ 38.9* (p<0.001) 37.8* (p<0.001) Porcentaje de reducción promedio desde el inicio (%) 17.6 ~ 37.2* (p<0.001) 35.6* (p<0.001) n=pacientes seleccionados de forma aleatoria que recibieron, por lo menos, una dosis del medicamento del estudio. La figura 1 muestra el porcentaje de pacientes (excluyendo los pacientes con levetiracetam concomitante) por categoría de reducción desde el inicio en la frecuencia de la crisis de inicio parcial por 28 días a través de los tres estudios. Los pacientes con más de un 25% de incremento en el padecimiento mencionado se muestran a la izquierda de “lo menos deseable”. Los pacientes con una mejoría en la reducción del procentaje desde el inicio en la frecuencia de dicho padecimiento se presentan en cuatro categorías a la derecha. Los porcentajes de pacientes con, al menos, un 50% de reducción en la frecuencia de las crisis fueron de 20.3%, 34.2%, 39.5% y 37.8% para el placebo, 50 mg/día, 100 mg/día, y 200 mg/día, respectivamente. Figura 1
La conclusión de que brivaracetam es efectivo como monoterapia en pacientes de 4 años y mayores se extrapoló a partir de los estudios controlados en epilepsia concomitante. Las simulaciones farmacocinéticas y farmacodinámicas mostraron que la dosis de brivaracetam, al ser utilizado como monoterapia, da como resultado una exposición y una exposición-respuesta que son similares a aquellas que han demostrado ser seguras y efectivas cuando se utiliza como terapia de adición para el tratamiento de las crisis de inicio parcial. Tratamiento con levetiracetam Aunque el número de pacientes es limitado, no se observó beneficio del brivaracetam contra el placebo en pacientes que toman levetiracetam de manera concurrente. No se observaron problemas de seguridad ni de tolerabilidad adicionales. En el estudio N01358, un análisis previamente especificado muestra que en pacientes con exposición previa al levetiracetam, se demostró la eficacia sobre el placebo en relación con dosis de 100 y 200 mg/día. Poblaciones especiales Población de edad avanzada Población pediátrica No se ha establecido la seguridad de la inyección de Briviact® en pacientes pediátricos. No se han establecido la eficacia y tolerabilidad de brivaracetam en pacientes menores de 4 años de edad. Estudios de extensión de etiqueta abierta
Hipersensibilidad al principio activo o a cualquiera de los excipientes. 7. PRECAUCIONES GENERALES
Ideación suicida y trastornos del comportamiento Los pacientes deben ser monitoreados para la detección de signos de ideas suicidas y trastornos del comportamiento y se debe considerar un tratamiento apropiado. Los pacientes (y sus cuidadores) deberán ser advertidos para buscar ayuda médica en caso de que se presenten signos de ideas suicidas o trastornos en el comportamiento. Insuficiencia hepática Los ajustes de dosis son recomendados en pacientes con insuficiencia hepática. Descontinuación Efectos sobre la capacidad para conducir y utilizar maquinaria. El tratamiento con brivaracetam ha sido asociado con somnolencia y otros síntomas relacionados con el sistema nervioso central (SNC). Por lo tanto, los pacientes deben ser advertidos de no conducir automóviles o de operar maquinarias potencialmente peligrosas hasta que se haya familiarizado con los efectos de brivaracetam sobre su habilidad de poder realizar tales actividades. Intolerancia a la Lactosa
Mujeres en edad reproductiva Embarazo Como una medida de precaución, brivaracetam no debe emplearse durante el embarazo, a menos que haya una clara necesidad de hacerlo (si el beneficio para la madre claramente sobrepasa el riesgo potencial para el feto). La suspensión de tratamientos antiepilépticos puede dar por resultado la exacerbación de la enfermedad que podría ser peligrosa para la madre y el feto. Si una mujer decide embarazarse, el uso del brivaracetam debe ser cuidadosamente reevaluado. Estudios realizados en animales no indicaron ningún efecto teratogénico en ratas o conejos. Trabajo de parto y parto Lactancia Fertilidad
Estudios clínicos • Resumen En todos los estudios controlados y no controlados en pacientes con epilepsia, 2388 sujetos han recibido el brivaracetam, de los cuales 1740 han recibido tratamiento durante > 6 meses, 1363 durante > 12 meses, 923 durante > 24 meses, 733 durante > 36 meses, y 569 durante > 60 meses (5 años). En los estudios controlados en la terapia adjunta con placebo en 1,558 pacientes con crisis de inicio parcial (1,099 de ellos tratados con brivaracetam y 459 con placebo), el 68.3% de los pacientes tratados con brivaracetam y el 62.1% de los tratados con placebo experimentaron eventos adversos. Las reacciones adversas más frecuentemente reportadas (> 10%) en los pacientes tratados con brivaracetam fueron: somnolencia 14.3% y mareo 11.0%. Estas por lo general se presentaron con una intensidad de leve a moderada. La somnolencia y la fatiga fueron reportadas con una mayor incidencia con el incremento de la dosis. Los tipos de reacciones adversas reportadas durante los primeros siete días del tratamiento fueron similares a las reportadas durante el período de tratamiento general. La tasa de descontinuación debido a reacciones adversas fue de 6.0%, 7.4% y 6.8% para los pacientes seleccionados de manera aleatoria para el brivaracetam en las dosis de 50, 100 y 200 mg/día, respectivamente y de 3.5% para pacientes seleccionados aleatoriamente con el placebo. La reacción adversa más frecuente que dio lugar a la suspensión de la terapia con brivaracetam fue mareo. La seguridad de brivaracetam como monoterapia en pacientes de 16 años y mayores con crisis de inicio parcial se extrapoló de los estudios controlados en epilepsia concomitante. Se espera que el uso de brivaracetam para la monoterapia tendría un perfil de reacciones adversas similares al de la terapia de adición. • Listado de Reacciones Adversas Las frecuencias se definen de la siguiente manera: muy frecuente (> 1/10), frecuente (> 1/100 a < 1/10), poco frecuente (> 1/1,000 a < 1/100). Dentro de cada uno de los grupos de frecuencia, los eventos no deseados se presentan en orden decreciente de seriedad. Infecciones e infestaciones Trastornos de la sangre y del sistema linfático Trastornos del metabolismo y de la nutrición Trastornos psiquiátricos Trastornos del sistema nervioso Trastornos respiratorios, torácicos y mediastínicos Trastornos gastrointestinales Trastornos generales y alteraciones en el lugar de administración Trastornos del sistema inmunológico Descripción de las reacciones adversas seleccionadas Se han reportado ideas suicidas en el 0.3% (3/1099) de pacientes con brivaracetam, y el 0.7% (3/459) de los pacientes con placebo. En los estudios clínicos de corto plazo sobre el fármaco mencionado en pacientes que padecen epilepsia, no se reportaron casos de suicidio ni de intento de suicidio, sin embargo, ambos se han reportado en los estudios de extensión de etiqueta abierta. Durante el desarrollo clínico han sido reportadas reacciones sugestivas de hipersensibilidad inmediata (tipo I) en un pequeño número de pacientes con brivaracetam (9/3022). Estudios de extensión de etiqueta abierta Pacientes pediátricos (4 años hasta menores de 16 años)
Los estudios de interacción únicamente se han realizado en adultos. Efectos de otros medicamentos sobre brivaracetam De esta forma, no es probable que la administración conjunta con inhibidores de CYP afecte de forma significativa la exposición de brivaracetam. Dicha administración conjunta con el inductor fuerte del CYP450, la rifampicina disminuye las concentraciones de brivaracetam en plasma en un 45%. Los médicos deben considerar el incremento de la dosis de este medicamento en pacientes que inician el tratamiento con rifampicina y disminuirlas cuando se suspende la terapia con rifampicina. Las concentraciones de brivaracetam no fueron modificadas representativamente por los inhibidores de CYP3A y de CYP2C19. Estudios in vitro mostraron que la disposición de este fármaco no debe afectarse de manera importante por ningún CYP (e.g., CYP1A, 2C8, 2C9, 2C19, 2D6 y 3A4) o inhibidores transportadores (por ejemplo, glicoproteína P, BCRP, MRPs). Efectos del brivaracetam sobre otros medicamentos Medicamentos antiepilépticos Tabla 2: Interacciones entre el brivaracetam y otros medicamentos antiepilépticos (AED). AED administrado en conjunto Influencia del AED sobre la concentración de brivaracetam en plasma Influencia de brivaracetam en la concentración de AED en plasmaCarbamazepina Disminución del 26% No se requiere ningún ajuste Ninguno Incremento de epóxido de carbamazepina (referirse a lo siguiente). No se requiere ajuste de dosis. Clobazam No hay datos disponibles Ninguna Clonazepam No hay datos disponibles Ninguna Lacosamida Sin datos Ninguno Lamotrigina Ninguna Ninguna Levetiracetam Ninguna Ninguna Oxcarbazepina Ninguna Ninguna (derivado de monohidroxi MHD) Fenobarbital Disminución del 19% No se requiere ajuste de dosis Ninguna Fenitoina Disminución del 21% No se requiere ajuste de dosis Ninguna Incremento del 20%* Pregabalina Sin datos Ninguna Topiramato Ninguna Ninguna Ácido valproico Ninguna Ninguna Zonisamida Sin datos Ninguna *basado en un estudio que implica la administración de una dosis supraterapéutica de 400 mg / día brivaracetam Además, los estudios in vitro mostraron que la disposición de brivaracetam no se ve afectada de forma significativa por el cannabidiol. Brivaracetam es un inhibidor reversible moderado de hidrolasa de epóxido que resulta de una concentración incrementada de epóxido de carbamazepina, un metabolito activo de carbamazepina. En estudios controlados, la concentración en plasma de epóxido de carbamazepina se incrementó en un promedio de 37%, 62% y 98% con una ligera variabilidad en las dosis del fármaco mencionado de 50, 100 y 200 mg/día, respectivamente. No se observó toxicidad. Anticonceptivos orales Cuando el brivaracetam fue coadministrado en una dosis de 400 mg/día (dos veces la dosis diaria mínima recomendada) con un anticonceptivo oral que contiene etinilestradiol (0.3 mg), y levonorgestrel (0.15 mg), se observó una reducción de las AUCs de estrógenos y progestina de 27% y 23%, respectivamente, sin impacto en la supresión de la ovulación, (no se observaron cambios en los marcadores endógenos: estradiol, progesterona, hormona luteinizante, hormona folículo estimulante y hormona sexual unida a la globulina). No se ha realizado ningún otro estudio con dosis inferiores de anticonceptivos orales. Otros El medicamento de este estudio incrementó el efecto de alcohol en la función psicomotora, atención y memoria en un estudio de interacción farmacocinética y farmacodinámica en sujetos sanos. No hubo interacción farmacocinética. 11. ALTERACIONES EN LOS RESULTADOS DE LAS PRUEBAS DE LABORATORIO
No se han reportado hasta el momento. 12. PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGENESIS, MUTAGENESIS, TERATOGENESIS Y SOBRE LA FERTILIDAD
Después de una administración aguda, se encontró que la dosis máxima oral no letal en ratas fue de ≥ 1000 mg/kg y se identificó un nivel sin efecto de 500 mg/kg (ambos sexos), con base en signos clínicos. En el paquete estándar de los estudios sobre la farmacología de seguridad, la evaluación del SNC, sistemas cardiaco, respiratorio y gastrointestinal mostraron que los efectos predominantes estuvieron relacionados con el SNC (principalmente la depresión transitoria del SNC, y se disminuyó la actividad locomotriz espontánea). Estos efectos fueron observados en múltiplos (mayores de 50 veces) de la dosis activa a nivel farmacológico de 2 mg/kg, principalmente desde 100 mg/kg. Además, el brivaracetam no afectó las funciones de aprendizaje ni de memoria como se muestra por lo ausencia de la alteración de la potenciación a largo plazo en porciones hipocámpicas de las ratas, y por la ausencia del efecto en la prueba del laberinto de agua de Morris y las ratas con la amígdala encendida hasta 21 mg/kg. No se observó ningún cambio hepático adverso en ratas y monos después de la administración crónica de brivaracetam en una exposición por encima (5 a 42 veces) de la exposición humana promedio, en la dosis clínica de 200 mg/día. En perros, la administración del medicamento citado produjo cambios adversos en el hígado, principalmente porfiria, en un nivel de exposición cercano a la exposición humana promedio en la dosis clínica de 200 mg/día. Sin embargo, los datos toxicológicos acumulados sobre brivaracetam y acerca de un compuesto relacionado en estructura indican que los cambios hepáticos de los perros se han desarrollado a través de los mecanismos no relevantes para humanos. La genotoxicidad se evaluó in vitro en células bacterianas y de mamíferos, e in vivo en ratas y ratones. Brivaracetam no mostró evidencia de la mutagenicidad o clastogenicidad. Se han realizado estudios sobre carcinogenicidad en ratas y ratones. Los hallazgos en ratas no indicaron ningún potencial oncogénico. Los hallazgos en ratones (modesto incremento en la incidencia de tumores hepatocelulares en ratones machos únicamente), se consideraron como el resultado de un mecanismo de acción no genotóxico ligado a una inducción enzimática hepática parecida a la fenobarbitona, un fenómeno específico conocido de los roedores. Brivaracetam no afectó la fertilidad de machos o hembras y ha demostrado que no tiene potencial teratogénico en ratas o conejos. Se observó embriotoxicidad en conejos a una dosis maternal tóxica de brivaracetam. Se demostró que brivaracetam en ratas, cruza fácilmente la barrera placentaria y que es excretado en la leche materna. Los efectos adversos potenciales de la administración oral a largo plazo del brivaracetam en el crecimiento y desarrollo neonatal se investigaron en ratas y perros jóvenes. En ratas jóvenes, la dosis más alta probada, 600 mg/kg/día, se consideró que inducía efectos adversos de desarrollo (por ejemplo, mortalidad, signos clínicos, disminución de peso corporal y reducción del peso cerebral). No se reportaron efectos adversos sobre la función del SNC, exámenes neuropatológicos e histopatológicos del cerebro. Se consideró que el NOAEL (nivel sin efecto adverso observable) era de 300 mg/kg/día. En perros jóvenes, la dosis de 100 mg/kg/día indujo cambios hepáticos adversos similares a los observados en animales adultos. No se reportaron efectos adversos en el crecimiento, densidad o fuerza ósea, evaluaciones del cerebro y del comportamiento neuronal, y evaluación de neuropatología. En el NOAEL se logró una exposición similar a brivaracetam en animales adultos y jóvenes, excepto en el día 4 posterior al nacimiento en donde se logró una exposición más alta en animales jóvenes en comparación con los adultos. El abuso potencial fue investigado en ratas y los estudios no indicaron un potencial en el abuso o dependencia. 13. DOSIS Y VÍA DE ADMINISTRACIÓN
Vía de administración: Oral. Información de la Dosificación Monoterapia o Terapia de Adición Tabla 1: Dosis recomendada para adultos y pacientes pediátricos de 4 años en adelante Edad y Peso Corporal Dosis Inicial Dosis de Mantenimiento Mínima y MáximaAdultos (16 años y mayores) 50 mg dos veces al día (100 mg al día) 25 mg a 100 mg dos veces al día (50 a 200 mg al día) Pacientes pediátricos con un peso de 50 Kg en adelante 25 mg a 50 mg dos veces al día (50 mg a 100 mg al día) 25 mg a 100 mg dos veces al día (50 a 200 mg al día) Pacientes pediátricos con un peso de 20 Kg y menor a 50 Kg 0.5 mg/Kg a 1 mg/Kg dos veces al día (1 mg/Kg a 2 mg/Kg al día) 0.5 mg/Kg a 2 mg/Kg dos veces al día (1 mg/Kg a 4 mg/Kg al día) Pacientes pediátricos con un peso de 11 Kg y menor a 20 Kg 0.5 mg/Kg a 1.25 mg/Kg dos veces al día (1 mg/Kg a 2.5 mg/Kg al día) 0.5 mg/Kg a 2.5 mg/Kg dos veces al día (1 mg/Kg a 5 mg/Kg al día) El uso de brivaracetam no se recomienda en menores de 4 años de edad, dado que su eficacia no se ha establecido todavía en esta población. Descontinuación Briviact® puede tomarse con o sin alimentos. La dosis diaria se administra en dos dosis iguales divididas, una vez en la mañana y otra en la noche. El tratamiento con Briviact® se puede iniciar por administración vía oral o intravenosa. Cuando la administración se convierta de oral a intravenosa o viceversa, deben mantenerse tanto la dosis diaria total como la frecuencia de la administración. La solución inyectable de Briviact® es una alternativa para los pacientes cuando la administración oral no es viable temporalmente. Las tabletas deben tomarse completas por vía oral y con líquido. Poblaciones especiales Pacientes de edad avanzada (a partir de 65 años) Insuficiencia renal Insuficiencia hepática
Para acceder a la información de posología en Vademecum.es debes conectarte con tu email y clave o registrarte. Conéctate Regístrate 14. MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL
Síntomas Manejo de la sobredosis
Caja de cartón con 14 tabletas con 10 mg en envase de burbuja e instructivo anexo.
No use Briviact® después de la fecha de caducidad que está impresa en la caja de cartón y en el blíster. Consérvese a no más de 30°C y en lugar seco. 17. LEYENDAS DE PROTECCIÓN
No se use en el embarazo, la lactancia, ni en menores de 4 años.
UCB de México, S.A. de C.V.
Reg. No. 379M2016 SSA IV
Mayo 2021 |